Genetic regulation & genome evolution

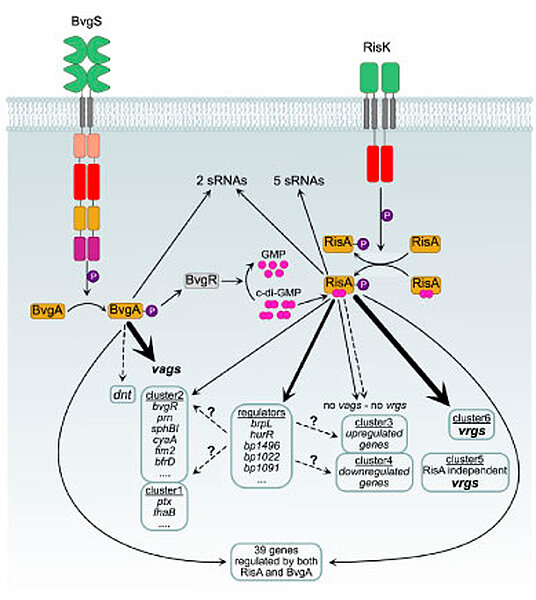
Bordetella virulence factor genes regulations
The Bordetellae, which include the human pathogen B. pertussis and the veterinary pathogen B. bronchiseptica, produce a range of virulence factors, including adhesins and toxins. These enable the bacteria to transmit, infect and colonize the respiratory tract of the host. These bacteria are able to regulate the expression of their virulence factors according to the environmental conditions they are faced with. The regulation of Bordetella virulence is mediated by the two-component system BvgA/S. During the virulence phase, the phosphorylation of BvgA activates the transcription of virulence-activated genes (vags). In the a-virulent phase, the vags are not expressed, but instead, virulence-repressed genes (vrgs) are expressed. The expression of the vrgs is regulated by another two-component system, RisA/K, and by the action of BvgR, a c-di-GMP phosphodiesterase. Our objective is to utilise molecular biology and omics technologies to ascertain the regulatory network and the mode of regulation involving the BvgASR and the RisAK systems. Notably, our research has revealed that certain genes are subject to regulation by both BvgA and RisA. Furthermore, it has been demonstrated that a number of genes which have not been found to be regulated in omics studies contain a binding site for BvgA and/or RisA in their promoter regions. This adds to the complexity of the RisAK/BvgASR network in Bordetella virulence regulation. This has enabled us to redefine the dogma of virulence regulation in Bordetella. We have identified new partners for the BvgAS/RisAK systems, which are under investigation using several omics approaches, as well as analysing protein-protein interactions and performing targeted mutagenesis. We are also defining the regulatory functions of RisA, particularly with regard to phosphorylation and binding of the cofactor c-di-GMP. Furthermore, it has been determined that B. pertussis is capable of encoding up to 16 different two-component systems, which lends further credence to the assertion that Bordetella gene expression biology is of an extremely complex nature.

Characterization of the emerging pathogen Bordetella holmesii
Human respiratory infections are experiencing an alarming resurgence, characterized by an increase in pseudo-pertussis syndromes. While Bordetella pertussis remains the predominant causative agent, a significant proportion of cases (>20-30%) are now attributable to Bordetella holmesii. This phylogenetically distinct bacterial species has a particular tropism for invasive systemic infections (bacteremia, pneumonia, septic arthritis, pericarditis), establishing itself as an emerging opportunistic pathogen that could eventually become a significant public health problem. Critically, current acellular vaccines, formulated against the immunodominant antigens of B. pertussis, do not confer any cross-immunity against B. holmesii, and its molecular virulence determinants remain largely unknown.
Furthermore, sensitivity to macrolides and third-generation cephalosporins varies between strains, complicating treatment. The emergence of this pathogen poses a widely underestimated health threat, requiring urgent characterization of its pathogenic mechanisms before it reaches a critical epidemic threshold. This issue requires an integrative approach combining comparative genomics, differential transcriptomics and functional analyses of secretion systems in order to elucidate its pathogenesis and develop molecular diagnostic tools and rational prophylactic strategies. With our internationally recognized expertise in Bordetella pathogenesis and our skills in the molecular biology of virulence systems, our team proposes to develop an integrated experimental model for B. holmesii. We will use our skills to identify and characterize the genetic determinants regulating the adhesion, invasion and haematogenous dissemination of this bacterium.
This project has been funded by the University of Lille Foundation (2026).

Genomics and evolution of drug resistance in M. tuberculosis
The global rise in antimicrobial resistance (AMR) underscores the critical need to target the mechanisms that limit the effectiveness of antibiotics, promote the development of resistance and lead to treatment failures. In addition to classical drug resistance, the effectiveness of antibiotics is also impaired by more complex, so-defined bacterial tolerance mechanisms, which do not increase the minimum concentration of a drug needed to prevent growth, but enhance bacterial survival to transient antibiotic exposure. These mechanisms are also believed to play a key role in the emergence of resistance. In ResTolTB, we aim to elucidate the role and dynamics of genetically encoded multidrug tolerance mechanisms in altering synergistic drug interactions and in the emergence of AMR in Mycobacterium tuberculosis, the deadliest infectious bacterium and the first contributor to mortality due to AMR. By gathering strong and complementary experience and multidisciplinary expertise, we use an ad hoc systemic approach combining genetics and experimental evolution in vitro and in vivo, in silico modeling, phylogenomics and GWAS analyses of circulating strains, as well as analyzes by multi-target deep sequencing and whole genome sequencing (GWS), to determine the evolutionary dynamics of tolerance/ persistence in evolved populations and clones, and to determine its role in the emergence of resistance. Using a new deep sequencing-based prototype test, we will also assess the detection and association of tolerance and resistance mutations, directly in clinical samples from patients on treatment for multidrug resistant or drug susceptible tuberculosis. The advanced understanding of the dynamic interactions between tolerance and resistance will allow the development of novel diagnostic and therapeutic tools.


